Pathologies plaquettaires
COMMISSION PATHOLOGIES PLAQUETTAIRES
![]() Plaquette pathologies plaquettaires (663.65 Ko)
Plaquette pathologies plaquettaires (663.65 Ko)
Qu’est qu’une plaquette ?
Les plaquettes sont des cellules sanguines, si petites qu’elles ont été ignorées pendant longtemps. Elles sont issues des mégacaryocytes (MK) qui sont des cellules qui prolifèrent et maturent dans la moelle osseuse. La fragmentation du cytoplasme des MKs donne naissance, au niveau des sinus vasculaires, aux plaquettes, cellules anucléées. Les plaquettes restent dans la circulation sanguine pendant 8 jours puis sont éliminées.
Quel est leur rôle ?
Leur principale mission est d’assurer l’arrêt des saignements lors de lésions vasculaires. En effet, la quantité de sang que peut perdre l’organisme est limitée. Pour se protéger de ce risque hémorragique les plaquettes colmatent les brèches des vaisseaux grâce à leurs propriétés adhésives qui vont se manifester aux sites de lésions.
Comment ces petites cellules colmatent les brèches ?
Les plaquettes possèdent à leur surface des molécules appelées récepteurs qui vont servir de point d’attache aux substances contenues dans la paroi vasculaire, comme le collagène ou le facteur Willebrand, rendues accessibles suite aux lésions des vaisseaux. Elles s’immobilisent progressivement aux niveaux des brèches vasculaires pour former une première couche encore bien fragile pour stopper le saignement. C’est alors qu’intervient une seconde série d’interactions entre protéines du plasma et récepteurs permettant la formation de plusieurs couches de plaquettes. C’est ainsi que se constitue l’agrégat plaquettaire beaucoup plus solide (figure 1). De plus, des travaux récents montrent que le rôle des plaquettes est certainement encore plus important, notamment au travers des molécules biologiquement actives qu’elles transportent et qui se déversent aux sites de la lésion. Ces substances favorisent la prolifération cellulaire et exercent ainsi un rôle dans la réparation tissulaire.
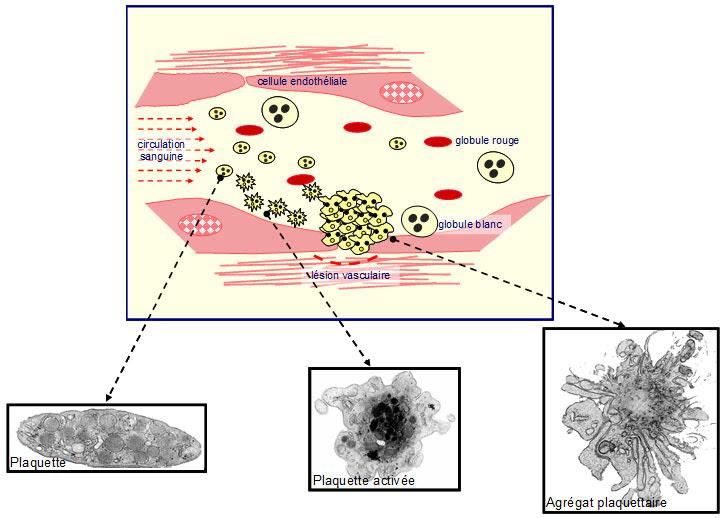
Figure 1 : Schéma montrant l’interaction des plaquettes (coloration jaune et rouge) au site vasculaire lésé.
L’apparition d’une brèche dans la paroi vasculaire va induire un saignement.
Les plaquettes sanguines vont assurer l’arrêt de ce saignement en colmatant cette brèche.
Les plaquettes à l’état normal sont de forme discoïde. L’activation plaquettaire entraîne un changement conformationnel de celles-ci, qui vont alors adhérer à la paroi lésée, puis s’agréger entre elles pour former un agrégat plaquettaire.
Les pathologies plaquettaires
Les thrombopénies et thrombopathies constitutionnelles correspondent à des déficits quantitatifs ou qualitatifs des plaquettes pouvant être à l’origine de saignements. Elles résultent d’anomalies génétiques. Ces pathologies peuvent être isolées ou bien associées à des anomalies affectant d’autres cellules sanguines ou des organes, on parle alors de pathologie syndromique.
Elles constituent un groupe de pathologies rares de mieux en mieux identifiées grâce à une meilleure connaissance des constituants des plaquettes et de la mégacaryocytopoïèse. Leur prévalence est estimée à 1/30 000.
Les maladies plaquettaires étudiées présentent une très grande diversité. Elles peuvent en fonction de la nature de l’anomalie se rassembler en différents groupes. Ci-dessous les principales étiologies.
LES PRINCIPALES THROMBOPATHIES :
Maladie de Glanzmann,
Déficit en CalDAG-GEF1,
Déficit en kindline3,
Déficit en récepteurs d’activation (P2Y12, etc.),
Déficit en granules (syndromes des plaquettes grises, déficit en granules denses).
LES PRINCIPALES THROMBOPÉNIES :
Thrombopénie par anomalie du cytosquelette (syndrome MYH9, anomalie ACTN1, etc.),
Thrombopénie par anomalie des facteurs de transcription (RUNX1, ETV6, GATA1, etc.),
Thrombopénie avec anomalie des granules (syndrome des plaquettes grises, etc.),
Thrombopénie avec anomalie des récepteurs de surface (syndrome Bernard-Soulier, etc.).
La présence d’une thrombopénie isolée et chronique chez un adulte et chez un enfant, fait classiquement évoquer en premier lieu un processus auto-immun en raison d’une plus forte prévalence. Le diagnostic différentiel avec une pathologie plaquettaire constitutionnelle génétique doit cependant être évoqué pour éviter la mise en place de traitements inappropriés et inefficaces. Plusieurs critères peuvent permettre de suspecter une thrombopénie d’origine génétique : la résistance aux traitements immunologiques, le contexte familial, l’histoire personnelle et familiale (anomalies associées), la consanguinité.
Les syndromes hémorragiques sont très variables en fonction de la pathologie plaquettaire concernée. Une bonne connaissance de la pathologie et la mise en oeuvre de mesures préventives doivent permettre de limiter le risque hémorragique.
Les pathologies plaquettaires : Parcours de soin
Les pathologies plaquettaires héréditaires sont des diagnostics difficiles. Celui-ci nécessite le recours à des médecins spécialistes appartenant au réseau du CRPP. Trente sites CRPP existent et sont répartis sur le territoire national et permettent de répondre totalement aux besoins des patients atteints de pathologies plaquettaires héréditaires.
Les objectifs du centre sont de dépister, diagnostiquer et prendre en charge les patients atteints de pathologies plaquettaires constitutionnelles.
Les Thrombopénies
Les thrombopénies sont des défauts plaquettaires d’ordre quantitatif. Leur connaissance a évolué ces dernières années et montre que certaines d’entre elles prédisposent à d’autres atteintes, altérant la qualité de vie voire sa durée. Les consultations en centres spécialisés (sites CRPP) vont permettre un diagnostic étiologique en fonction des résultats de l’enquête clinique, biologique et génétique. La connaissance de l’étiologie permettra d’évaluer le pronostic, de personnaliser le suivi et d’adapter la prise en charge.
Plusieurs étiologies vont donner lieu à des thrombopénies modérées dont certaines resteront stables et ne s’accompagneront pas de risque particulier. Pour ces individus, l’exploration aura l’avantage d’apporter une certitude diagnostique et d’éviter la mise en place de traitements inefficaces.
Les Thrombopathies
Les thrombopathies héréditaires correspondent à des défauts fonctionnels. Elles sont isolées ou associées à une thrombopénie. Le syndrome hémorragique est au premier plan. Il est nécessaire de reconnaître les formes sévères qui ont besoin d’une prévention du risque hémorragique en cas d’urgence ou de chirurgie programmée, basée sur la prescription de concentrés plaquettaires ou de facteur VII activé. Parmi ces pathologies, la maladie de Glanzmann est la mieux décrite. Le diagnostic des thrombopathies repose sur l’exploration fonctionnelle des plaquettes, qui nécessite le recours à des laboratoires spécialisés adossés aux sites du CRPP.
Les difficultés de diagnostic et la multidisciplinarité des compétences techniques requises nécessitent une étroite collaboration entre spécialistes. L’établissement d’un diagnostic nécessite en général deux à trois consultations. Le niveau des explorations s’incrémente au regard des explorations précédentes. Les dossiers sont discutés en réunions médicales et pour les cas compliqués en réunions de concertation pluridisciplinaires nationale (visioconférence). Dans tous les cas, le médecin traitant est informé en diagnostic et la prise en charge à mettre en œuvre.
Pathologies plaquettaires : Thérapeutique
Les traitements des hémorragies sont les traitements symptomatiques usuels (anti-fibrinolytiques, traitement hormonal des saignements gynécologiques, hémostatiques d’appoint, traitements de la carence en fer).
Lorsque le saignement ne peut être contrôlé par ces moyens, le seul recours est l’administration préventive ou curative de concentrés plaquettaires. Leur efficacité est excellente dans la majorité des cas, mais une minorité de patients peuvent développer des anticorps contre des antigènes plaquettaires, qui réduisent l’amplitude et la durée de l’effet des plaquettes transfusées. Leur sécurité et leur tolérance sont très bonnes, en dehors de possibles réactions transfusionnelles de faible intensité et facilement contrôlables. Le facteur VII activé (novoseven) est utile dans certains cas pour traiter ou prévenir le saignement.
La recherche thérapeutique concerne les médicaments analogues de l’hormone thrombopoïétine (romiplostim par voie sous-cutanée, eltrombopag par voie orale) qui stimulent la production des plaquettes par les mégacaryocytes. Ces médicaments, utilisés pour le traitement de certaines thrombopénies acquises, semblent pouvoir atténuer la symptomatologie hémorragique spontanée et corriger la thrombopénie, au moins transitoirement, mais leur bénéfice et leur sécurité doivent être confortés et leur utilisation n’est pas actuellement autorisée dans les thrombopénies constitutionnelles, hors essais cliniques. Enfin, la thérapie cellulaire (allogreffe de cellules-souches hématopoïétiques), ne s’adresse qu’à de rares formes sévères notamment celles associées à un déficit immunitaire mettant en jeu le pronostic vital.
https://www.youtube.com/watch?v=tkXcbJb_Nys&ab_channel=CRPPMarseille